Fenilketonurija - klasični znaki, kot dedna in prehranska terapija
Vsebina
- 1Kako se manifestira bolezen fenilketonurije
- 2Mehanizem razvoja bolezni
- 3Fenilketonurija pri otrocih
- 4Simptomi bolezni
- 5Vzroki in provokativni dejavniki
- 6Diagnostika
- 7Zdravljenje klasične fenilketonurije
- 8Prehrana novorojenčkov in dietna terapija
- 9Prehrana za predšolske otroke in šolarje
- 10Skupine izdelkov na PKU
- 11Kako nadzorovati raven fenilalanina v krvi
- 12Videi
Bolezen, katere pojav je povezan z okvarami genetske celične naprave - fenilketonurija - je vključen v majhen seznam dednih bolezni, ki se zdravijo. Odkritje te bolezni je bil norveški zdravnik IA Felling, kasneje pa je bilo ugotovljeno, da razvoj in potek bolezni ustreza enemu genu, ki se imenuje gen fenilalaninske hidroksilaze (dolgo ramo 12. kromosoma, ki vsebuje do 4,5% celotnega celičnega DNA celice). Podedovana napaka vodi do delne ali popolne deaktivacije encima jeter fenilalanin-4-hidroksilaze.
Kako se manifestira bolezen fenilketonurije
Dedna bolezen fenilketonurije (PKU) vodi v kronično zastrupitev telesa s strupenimi snovmi, ki nastanejo zaradi oslabljene presnove aminokislin in procesa.hidroksilacijo fenilalanina. Trajna zastrupitev povzroča poškodbe centralnega živčnega sistema (CNS), katerih izrazna značilnost je postopno zmanjšanje inteligence (fenilpiruvična oligofrenija).
Bolečina sečnje se kaže v čezmernem kopičenju fenilalanina v telesu in v proizvodih neprimerne presnove. Drugi dejavniki razvoja fenilketonurije vključujejo zdrobljen transport aminokislin skozi krvno-možgansko pregrado, nizke ravni nevrotransmiterjev (serotonin, histamin, dopamin). V odsotnosti pravočasno zdravljenje bolezni vodi v duševno zaostalost in lahko povzroči smrt otroka.

Mehanizem razvoja bolezni
Vzročni dejavnik pri pojavu genskih motenj je presnovni blok, ki moti tvorbo fenilalanin-4-hidroksilaze (encima, ki je odgovoren za pretvorbo amino kisline fenilanina v tirozin). Proteinogena aminokislina tirozin je sestavni del beljakovin in pigmenta melanina, zato je nujen element za delovanje vseh telesnih sistemov, pomanjkanje pa vodi do fermentopatije.
Inhibicija metabolita, ki je posledica mutacijske inaktivacije encima, je aktivacija pomožnih poti za izmenjavo fenilalanina. Aromatska alfa-aminokislina se zaradi pomanjkljivih procesov izmenjave razgradi v toksične derivate, ki se ne tvorijo v normalnih pogojih:
- fenilpiruvična kislina (fenilpiruvat) - maščobni aromatski alfa-ketoacid, njeno izobraževanjevodi do mielinacije procesov nevronov in demence;
- fenilmaktična kislina - produkt, ki nastane med regeneracijo fenil virionske kisline;
- Feniletilamin - začetna spojina za biološko aktivne oddajnike elektrokemijskih impulzov, poveča koncentracijo dopamina, adrenalina in noradrenalina;
- Ortofenilacetat - strupena snov, ki povzroča kršitev presnovnih procesov fekalnih spojin v možganih.
Medicinske statistike kažejo, da je patološko spremenjeni gen prisoten pri 2% prebivalstva, vendar se ne kaže. Genetska okvara se prenaša na otroka od staršev le ob prisotnosti bolezni pri obeh partnerjih, pri čemer otrok v 50% primerov postane nosilec mutiranega gena, ki ostaja zdrav. Verjetnost, da bo fenilketonurija pri novorojenčkih privedla do bolezni, je 25%.
Katera vrsta je podedovana
Bolezen pri sečnji je genetsko odstopanje, natisnjeno na avtosomno recesivno vrsto. Ta vrsta dedovanja pomeni, da se razvoj znakov prirojene bolezni pojavi le, če je en otrok podedovan iz poškodovane genetske kopije obeh staršev, ki sta heterozigotni nosilci spremenjenega gena.
Razvoj kongenitalne bolezni v 99% primerov povzroča mutacijo gena, odgovornega za encimsko kodiranje, ki zagotavlja sintezo fenilalanin-4-hidroksilaze (klasična fenilketonurija). Do 1% genetskih bolezni je povezanih z mutacijskimi spremembami, ki se pojavljajo v drugih povzročiteljih genovpomanjkanje dihidropteridin reduktaze (tipa II PKU) ali tetrahidrobiopterina (PKU tipa III).
Fenilketonurija pri otrocih
Klasična oblika genitalnih bolezni pri otrocih se v večini primerov kaže od zunaj opaznih znakov od 3-9 mesecev življenja. Novorojenčki z okvarjenim genom izgledajo zdravo, posebnost pa je posebna navada (videz) otroka. Simptom se pojavi 6-12 mesecev po rojstvu.
Za PKU tipa II je značilno, da se prvi klinični simptomi pojavijo po 1,5 letih od nastopa svetlobe. Znaki bolezni ne izginejo po diagnosticiranju genetskih nepravilnosti in začetku prehranske terapije. Ta vrsta prirojene bolezni pogosto vodi do usodnega izida 2-3 let življenja otroka. Najpogostejši simptomi tipa II PKU so:
- izražena odstopanja v duševnem razvoju;
- hiperrefleksija;
- kršitev motoričnih funkcij vseh okončin;
- sindrom nenadzorovanih mišičnih kontrakcij.
Klinični znaki mutacijskih sprememb v genih tipa III so podobni boleznim, ki se pojavljajo pri tipu II. Za pomanjkanje tetrahidrobiopterina je značilna triada posebnih simptomov:
- visoka stopnja duševne zaostalosti;
- navidezno zmanjšana velikost lobanje glede na druge dele telesa;
- spastičnost mišic (z možnostjo popolne izgube gibanja uda).

Manifestacije bolezni sečnje
Med kliničnimi študijami in opažanji je bilo predlagano, da je učinekstrupenih derivatov izmenjave fenilalanina povzroči zmanjšanje intelektualnih sposobnosti, ki je progresivno in lahko privede do demence (oligofrenija, idioti). Med verjetnimi vzroki nepopravljivih kršitev možganske aktivnosti je najbolj smiselno, da se pripiše zmanjšanju ravni tirozina, pomanjkanju nevrotransmiterjev, ki prenašajo impulze med nevroni.
Natančen vzročno-posledični odnos med dednimi boleznimi in možganskimi motnjami do sedaj ni bil razkrit, kot je mehanizem razvoja zaradi fenilketonurije takih duševnih stanj kot ehpraxia, echolalia, divji napadi in razdražljivost. Rezultati analiz kažejo, da ima fenilalanin neposreden toksični učinek na možgane, kar lahko povzroči tudi zmanjšanje inteligence.
Posebnosti strukture in fenotipa
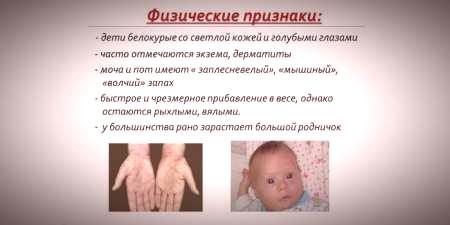
Glede na to, da je nasičenost pigmenta kože in las odvisna od ravni tirozina v mitohondrijih hepatocitov in da fenilketonurija povzroči ustavitev pretvorbe fenilalanina, imajo bolniki s to boleznijo fenotipske značilnosti (recesivni znaki). Povečan mišični tonus povzroča pojav odstopanj v strukturi telesa - postane displastičen. Odlične zunanje lastnosti fenilketonurije vključujejo:
- hipopigmentacija - svetla koža, svetlo modre oči, razbarvane lase;
- cinizem okončin;
- zmanjšana velikost glave;
- specifičen položaj telesa - ko poskušate stati ali sedeti, otrok vzame držo "krojača" (roke in noge, ki so zgibane v sklepih).
Simptomibolezen
Pravočasno se je Fellingova bolezen uspešno zdravila s prilagajanjem prehrane in razvoj otroka se izvaja v skladu z njegovo starostno skupino. Težava pri odkrivanju genske mutacije je v tem, da je za zgodnje znake težko odkriti celo izkušenega pediatra. Resnost simptomov prirojenih bolezni se poveča, ko otrok postaja starejši, saj uporaba beljakovinskih živil prispeva k razvoju motenj centralnega živčevja.
Znaki pri novorojenčkih
V prvih dneh otrokovega življenja je težko odkriti znake patoloških nenormalnosti - otrok se naravno obnaša, razvoj ni zamude. Simptomi bolezni se najprej pojavijo v 2 do 6 mesecih po rojstvu. Starši morajo biti pozorni na obnašanje otroka, za katerega je značilna nizka aktivnost, letargija ali, nasprotno, tesnoba, hiperboličnost.
Z začetkom dojenja novi beljakovini začnejo vstopati v telo novorojenčka z mlekom, ki služi kot katalizator za pojav prvih znakov, kar jasno kaže, da se je bolezen začela razvijati. Posebne klinične manifestacije bolezni so:
- stalno bruhanje (pogosto za prirojeno zoženje vratarja);
- pogoste motnje;
- brez odziva na zunanje dražljaje;
- mišična distonija (zmanjšana napetost mišic);
- konvulzivni sindrom (epileptični ali neepileptični napadi).
Simptomi pri otrocih po 6 mesecih
Če manifestacija genetske bolezni niv prvih 6 mesecih po rojstvu otroka se je pojavilo (ali ni bilo opaženo), potem pa po tem obdobju že lahko natančno določite zaostanek psihomotoričnega razvoja. Simptomi genetskih motenj, ki jih povzroča pomanjkanje encimov pri otrocih, starejših od šestih mesecev, so:
- zmanjšanje aktivnosti (do popolne brezbrižnosti);
- pomanjkanje poskusov vstajanja, sedenje;
- poseben "mišji" vonj kože (vonj plesni nastane zaradi umika toksičnih derivatov fenilalanina preko žlez znoj in urina);
- izguba sposobnosti vizualizacije obrazov staršev;
- lupljenje kože;
- pojav dermatitisa, ekcema, skleroderme.
Napredovanje bolezni v odsotnosti zdravljenja v otroštvu
Če v otroštvu niso odkrili razvojnih odstopanj in ni bilo opravljeno ustrezno zdravljenje, bolezen začne aktivno napredovati in pogosto vodi do invalidnosti. Pomanjkanje terapije v zgodnji fazi bolezni povzroča nastanek takšnih simptomov bolezni pri starosti 1,5 let:
- mikrocefalija (zmanjšana velikost možganov);
- odtok (premik vrha zgornjega zoba naprej);
- poznejši izbruh zob;
- hipoplazija sklenine (tankost ali popolna odsotnost zobne sklenine);
- zavlačevanje jezikovnega razvoja do popolnega pomanjkanja govora;
- 3, 4 stopnja oligofrenije (duševna zaostalost, duševna zaostalost);
- prirojena srčna bolezen (napake v strukturi srčne mišice, srce,velika plovila);
- motnje vegetativnega sistema (akrocijanoza, povečano znojenje, arterijska hipotenzija);
- zaprtje.
Vzroki in provokativni dejavniki
Da bi se manifestirala mutacija z avtosomno recesivno dedovanje, mora biti poškodovan gen podedovan od obeh staršev. Takšne genetske bolezni najdemo pri enaki pogostnosti pri novorojenčkih in dekletih. Patogeneza PKU je posledica kršitve izmenjave fenilalanina, ki se lahko pojavi v treh oblikah. Zdravljenje z dietno terapijo je predmet klasične fenilketonurije tipa I.
Atipične oblike bolezni se lahko pozdravijo s prilagoditvijo prehrane. Ta odstopanja so posledica pomanjkanja tetrahidropterina, dehidropterin reduktaze (redko - pirvatetetrahidropterinstanza, gvanozin-5-trifosfat ciklohidrolaze itd.). Večina primerov smrtnih primerov je bila zabeležena pri bolnikih z redkimi variacijami PKU, pri čemer so klinične manifestacije vseh oblik bolezni podobne. Tveganje za porod s mutiranim genom fenilalanin hidroksilaze se poveča, če so starši bližnji sorodniki (s tesno povezanimi poroki).
Diagnostika
V primeru suma na genetsko motnjo se diagnoza ugotovi na podlagi podatkov, pridobljenih na podlagi študije anamneze bolezni - genealoških podatkov, rezultatov kliničnih in medicinskih genetskih raziskav. Za pravočasno odkrivanje prirojenih bolezni (PKU, cistična fibroza, galaktozemija itd.) Program obvezne maseLaboratorijski pregled vseh novorojenčkov (neonatalni presejalni testi).
Če se bodo prihodnji starši zavedali prisotnosti mutiranega gena, sodobna medicina ponuja načine za odkrivanje napake med nosečnostjo (prenatalna diagnoza fetusa z invazivno metodo). Za delitev fenilketonurije po vrsti resnosti se uporablja pogojna razvrstitev glede na raven fenilalanina v fibrinogeni tekočini, ki izhaja iz plazemske plazme:
- Težka fenilketonurija - 1200 mkmol /l.
- Povprečno - 60-1200 μmol /l.
- Enostavno (ni potrebna obdelava) - 480 μmol /l.
Presejalni test
Zaznavanje genetskih nepravilnosti poteka v več fazah. Na prvi stopnji v bolnišnici pri vseh dojenčkih se opravi 3-5 dni življenja, vzorčenje periferne krvi (od petih) za raziskave. Material se nanaša na papir in se pošlje v biokemični laboratorij, kjer poteka njegova biokemična analiza. V drugi fazi presejalnega testa se določi koncentracija fenilalanina normalne vrednosti.

Če se patološke spremembe ne odkrijejo, se diagnoza konča, kar se zabeleži v otroški kartici. Ob odstopanju od norme se rezultati diagnoze pošljejo zdravniku-pediatru, da opravi natančnejšo študijo vzorca krvi novorojenčka. Zdravje otroka je odvisno od pravočasnega in natančnega izvajanja vseh ukrepov za odkrivanje nepravilnosti. Če se diagnoza potrdi po ponovnem presejalnem testu, bodo otrokovi staršikliniko za otroško genetiko za namene zdravljenja.
Analize in študije za potrditev diagnoze
Ponovna diagnoza v primeru odkritja odstopanja od norme med začetnim presejalnim testom se izvede s ponovno oceno analiz. Poleg določanja vsebnosti fenilalanina v krvi za diagnozo PKU pri otrocih in odraslih vključujejo:
- Preskus sečnje - določanje fenilpiruvične kisline v urinu z dodajanjem železovega klorida biomaterialu (obstaja modro-zelena obarvanost);
- Gatree test - ocena stopnje reakcije mikroorganizmov na presnovne produkte ali encime, ki jih vsebuje bolnikova kri;
- kromatografija - preučevanje kemijskih lastnosti snovi, razdeljenih med dve fazi;
- fluorimetrija - obsevanje biomateriala z monokromatskim sevanjem za določanje koncentracije snovi, ki jih vsebuje;
- elektroencefalografija - diagnoza električne aktivnosti možganov;
- slikanje z magnetno resonanco - kršitev atomskih jeder celic z elektromagnetnimi valovi in merjenje njihovega odziva.
Zdravljenje klasične fenilketonurije
Temelj zdravljenja s fenilketonurijo je omejitev porabe proizvodov, ki je vir beljakovin živalskega in rastlinskega izvora. Edini način uspešnega zdravljenja je dietna terapija, katere ustreznost se ocenjuje z vsebnostjo fenilalanina v serumu. Najvišja dovoljena raven aminokislin pri bolnikih različnih starostnih skupinje:
- pri novorojenčkih in otrocih, mlajših od 3 let - do 242 μmol /l;
- za predšolske otroke - do 360 mkmol /l;
- pri bolnikih, starih od 7 do 14 let - do 480 μmol /L;
- pri mladostnikih - do 600 μmol /l.
Učinek prehrane je odvisen od tega, v kateri fazi bolezni gre za popravek prehrane. Pri zgodnji diagnozi prirojene patologije je predpisana prehranska terapija od 8 tednov življenja (po tem obdobju se že začnejo nepovratne spremembe). Pomanjkanje pravočasnih ukrepov povzroči zaplete in zmanjšanje ravni inteligence za 4 točke v enem mesecu od rojstva do začetka zdravljenja.

Glede na to, da terapevtska prehrana za fenilketonurijo vključuje popolno izključitev iz prehrane živalskih beljakovin, je treba uporabiti druge vire esencialnih aminokislin kot tudi vitamine skupine B, mineralne spojine, ki vsebujejo kalcij in fosfor. Izdelki, ki naj bi se dodali prehrani brez beljakovin, vključujejo:
- beljakovinski hidrolizati (amigeni, aminazol, fibrinoze);
- ne vsebujejo zmesi fenilalanina, nasičenih z esencialnimi aminokislinami - tetrafenom, brez fenila.
Skupaj s kurativnimi ukrepi za odpravo vzroka za delovanje organizma je treba izvajati simptomatsko zdravljenje, namenjeno odpravljanju govornih okvar in normalizaciji koordinacije gibov. Kompleksna terapija vključuje fizioterapevtske postopke, masažo, pomoč terapevta, psihologa, izvajanje gimnastičnih vaj. V nekaterih primerih z dietno terapijokaže uporabo antikonvulzivov, nootropnih in žilnih zdravil.
Značilnosti zdravljenja atipičnih oblik
Fenilketonurija tipa II in III se ne more zdraviti z dieto z nizko vsebnostjo beljakovin - raven fenilalanina v krvi ostaja nespremenjena pri omejevanju pretoka beljakovin v telesu ali pa se klinični simptomi izboljšajo, tudi če se raven amino kisline zmanjša. Učinkovito zdravljenje teh oblik bolezni se izvaja z uporabo:
- tetrahidrobiopterin - dejavnik prizadetega encima;
- sintetični analogi tetrahidrobiopterina - te snovi bolje prodrejo skozi krvno-možgansko pregrado;
- zdravila za nadomestno zdravljenje - ne odpravljajo vzroka fenilketonurije, temveč podpirajo normalno delovanje telesa (levodopa v povezavi s karbidopo, 5-oksitriptofanom, 5-formiltetrahidrofolatom);
- hepatoprotektorji - podpirajo delovanje jeter;
- antikonvulzivi;
- uvedba gena fenilalanin hidroksilaze v jetra - eksperimentalna metoda.
Značilnosti prehrane novorojenčkov in terapija z dieto
V prvem letu življenja otroka je dovoljeno dojiti, vendar je treba njegovo količino omejiti. Do 6 mesecev je sprejemljiva raven porabe fenilalanina 60-90 mg na 1 kg telesne teže (100 g mleka vsebuje 5,6 mg fenilalanina). Od 3 mesecev je treba otrokovo prehrano postopoma razširiti z uvedbo sadnih sokov in pire krompirja.
Za otroke, starejše od 6 mesecev, je dovoljen vnos rastlinskega pireja, kaše (iz saga), brez beljakovin, \ tpoljubov Po 7 mesecih lahko otroku pripravite testenine z nizko vsebnostjo beljakovin, z 8 meseci - kruha brez beljakovin. Starost, pri kateri je treba omejiti prenos beljakovin v telesu bolnega otroka, ni določena. Zdravniki še vedno razpravljajo o vprašanju primernosti prehranske terapije, vendar se strinjajo, da je treba spoštovati vsaj 18 let, prehransko prehrano.
Fenilketonurija, diagnosticirana z žensko, ni razlog za opustitev rojstva otroka. Prihodnje mame s PKU za preprečevanje okvare ploda med nosečnostjo in za preprečevanje morebitnih zapletov so potrebne pred načrtovanim zanosom in med nošenjem otroka, da se drži diete z omejevanjem fenilalanina (njegova koncentracija v krvi mora biti do 242 mikromolov /liter).
Zmesi cepiv za dojenčke
Prehrana s fenilketonurijo temelji na znatnem zmanjšanju odmerka naravnih beljakovin v dnevni prehrani, vendar se telo novorojenčka ne more normalno razviti, če ni potrebnih elementov v sledovih. Da bi zapolnili otrokovo potrebo po beljakovinah, se uporabljajo aminokisline, ki ne vsebujejo antibiotikov in ki morajo biti v skladu z rusko zakonodajo brezplačne.
Otrokova toleranca na fenilalanin v prvem letu življenja se hitro spreminja, zato je treba nadzorovati njegovo koncentracijo v krvi otroka in prilagoditi prehrano. Mešanice so za določene starostne skupine:
- za dojenčke do leta imenovani Affenilak 15, Analog-JV, PKU-1, PKU-mix, PKU Anamix;
- za otroke, starejše od 1 letaleto, predpisati obogaten z vitamini in minerali mešanico z visoko vsebnostjo beljakovin - PKU Prima, P-AM Universal, PKU-1, PKU-2, HRM Maxamed, HR Maxumum.

Prehranski proizvodi za dopolnitev zalog beljakovin
Ena glavnih sestavin prehranske diete za fenilketonurijo so živila z nizko vsebnostjo beljakovin na osnovi škroba. Ti dodatki vsebujejo hidrolizat kazein, triptofan, tirozin, metionin, dušik in zagotavljajo otrokovo dnevno potrebo po otrokovih beljakovinah, kar je bistveno za normalen razvoj in rast. Specializirani izdelki, ki zapolnjujejo pomanjkanje potrebnih mineralov in aminokislin v pomanjkanju prehrane, so:
- Berlofen;
- Tsimorgan;
- Minafen;
- Apotoni.
Prehrana za predšolske otroke in šolarje
Ker se telo prilagodi na fenilalanin pri otrocih, starejših od 5 let, je mogoče postopno zmanjšati prehranske omejitve. Razširitev prehrane je uvedba žit, mlečnih izdelkov, mesnih izdelkov. Srednješolci že imajo visoko toleranco na fenilalanin, zato lahko v tej starosti še naprej razširjate prehrano, medtem ko je potrebno spremljati odziv na vse spremembe v prehrani. Za spremljanje otrokovega stanja se uporabljajo naslednje metode:
- ocena nevroloških parametrov, psihološkega stanja;
- nadzor parametrov elektroencefalograma;
- Določanje ravni fenilalanina.
Skupine izdelkov na PKU
V prehrani bolnikov s PKU skupaj z ne-beljakovinamiŠkrobni izdelki in terapevtske mešanice vključujejo tudi proizvode naravnega izvora. Pri pripravi menija je treba jasno izračunati količino zaužite beljakovine in ne preseči priporočenega odmerka zdravnika. Za izključitev toksičnih učinkov na telo se razvijejo 3 seznami proizvodov, ki vsebujejo prepovedane (rdeče), nereprezentirane (oranžne) in dovoljene (zelene) položaje.
Rdeči seznam
Fenilketonurija se razvije na podlagi odsotnosti encima, ki se pretvori v tirozin fenilalanin, zato je visoka vsebnost beljakovin osnova za dodelitev proizvodov na prepovedan (rdeči) seznam. Položaji s tega seznama bi morali popolnoma izključiti prehrano bolnika na PKU:
- meso;
- notranji organi živali, stranski proizvodi;
- klobase, klobase;
- morski sadeži (vključno z ribami);
- jajca vseh ptic;
- kislo mlečni izdelki;
- matice;
- stročnice in žitarice;
- izdelki iz soje;
- želatinaste jedi;
- slaščice;
- aspartam.
Oranžni seznam
Izdelki, ki se vnašajo v telo otroka z diagnozo PKU, so vključeni v oranžni seznam. Vključitev v prehrano predmetov s tega seznama je dovoljena, vendar v strogo omejenih količinah. Čeprav ti izdelki ne vsebujejo veliko beljakovin, lahko pa tudi povečam raven fenilalanina, zato njihova uporaba ni priporočljiva:
- zelenjava v konzervah;
- jedi iz krompirja in riža;
- zelje;
- mleko;
- šerbet.
Zeleni seznam
Zdravila, ki niso beljakovinska, so dovoljena za uporabo pri bolnikih s fenilketonurijo brez kakršnih koli omejitev. Pred nakupom izdelkov iz zelenega seznama morate pregledati sestavo, navedeno na embalaži, in se prepričati, da ni fenilalanina, ki vsebuje barvilo aspartama:
- sadje;
- zelenjava (razen krompirja in zelja);
- jagode;
- zelenice;
- škrobne žitarice (sago);
- med, sladkor, marmelada;
- moke iz moke iz koruze ali riževe moke;
- maslo, maščobe (maslo, sončnice, oljke).
Kako nadzorovati raven fenilalanina v krvi
Fenilketonurija je neozdravljiva bolezen, ki se lahko spremeni v fazo stagnacije z uporabo dietne terapije ter terapevtskih in profilaktičnih ukrepov. Ob spremembi življenjskih pogojev, kršitvi prehrane, se lahko bolezen zopet poslabša, zato bolniki potrebujejo doživljenjsko opazovanje. Kontrolni postopek je periodično določanje ravni fenilalanina v krvi. Pogostost dostave je odvisna od starosti bolnika:
- do 3 mesece - pregled je treba opraviti tedensko, dokler se ne dosežejo stabilni rezultati;
- od 3 mesecev do 1 leta - 1-2 krat na mesec;
- od 1 do 3 let - 1krat v 2 mesecih;
- več kot 3 leta - četrtletno.

Zdi se, da je kri za analize 3-4 ure po jedi. Poleg presejalnih pregledov se razvoj PKU nadzoruje z določanjem prehranskega statusa, fizičnega, čustvenega razvoja bolnika, ravni intelektualnih sposobnostiin razvoja jezika. Glede na rezultate opazovanj je lahko potrebna dodatna diagnostika ob sodelovanju ustreznih strokovnjakov.
Video posnetki
Informacije v članku so informativne narave. Materiali članka ne zahtevajo neodvisne obravnave. Le kvalificiran zdravnik lahko diagnosticira in svetuje glede zdravljenja na podlagi posameznih značilnosti posameznega bolnika.















